Forscher entdecken Ursachen für seltenes Syndrom
Veränderungen im MSL3-Gen führen zu Nerven- und Entwicklungsstörungen bei Kindern
Die Erforschung seltener Erkrankungen hat durch neue Verfahren der Hochdurchsatz-Sequenzierung sowie Plattformen zum Austausch zwischen Forschern und behandelnden Ärzten in den letzten Jahren einen entscheidenden Schub erhalten. In einer Kooperation mit französischen Kollegen der Universität Dijon sowie Ärzten weltweit ist es einem Team um Max-Planck-Direktorin Asifa Akhtar am MPI für Immunbiologie und Epigenetik gelungen, die Ursachen eines seltenen Syndroms aufzuklären. Bei diesem leiden betroffene Kinder an schweren Entwicklungsverzögerungen sowie fortschreitenden neurologischen Fehlfunktionen. Die Forscher konnten zeigen, dass Mutationen im Gen MSL3 zu einer Fehlregulation epigenetischer Mechanismen führen, die zentrale Gene während der Embryonalentwicklung steuerten. In der Studie, die in der Fachzeitschrift Nature Genetics erschien, konnte darüberhinaus in der Zellkultur ein vielversprechenden Weg für zukünftige epigenetische Therapien aufgezeigt werden, der Verlauf und Schwere des Syndroms bei den Patienten mildern könnte.
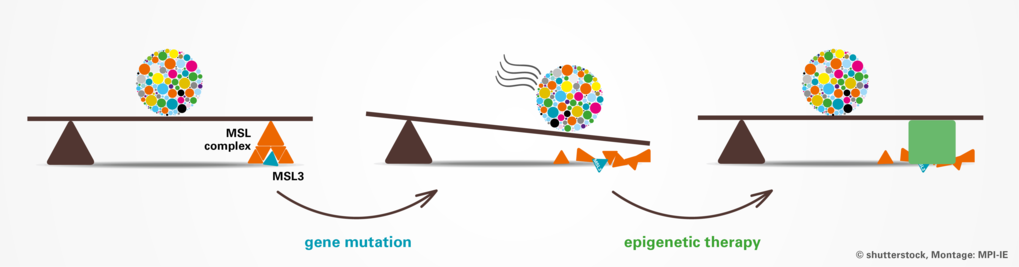
 führen zu einer Fehlregulation der enzymatischen Aktivität des MSL-Komplexes. Dieser ist für die Acetylierung des Lysins an Position 16 des Histon 4 und epigenetische Modifikationen verantwortlich ist. Die beeinträchtigte Funktion dieses epigenetischen Regulators führt bei Patienten zu einer Verringerung der Aktivität der wichtigsten Entwicklungsgene (bunte, gepunktete Kugel). Durch die Verwendung von Histondeacetylase-Inhibitoren (grünes Rechteck) konnte die kompromittierte Funktion des MSL-Komplexes in Zellen der Patienten wiederhergestellt werden.")
Die Ära der Postgenomik hat die Diagnose von bisher mysteriösen Symptomen und Krankheiten revolutioniert. Durch die neuen Verfahren der Hochdurchsatz-Sequenzierung ist es möglich geworden, unzählige neue Mutationen im Zusammenhang mit menschlichen Krankheiten zu identifizieren. Dies hat einen Wendepunkt gerade für die sogenannten „Waisen der Medizin“ geschaffen: die seltenen Erkrankungen, also Krankheiten, von denen nur wenige hundert Menschen weltweit betroffen sind. In vielen Fällen zeigen betroffene Patienten sehr komplexe und vielfältige Symptome und haben derzeit meist keine bis wenig Behandlungsmöglichkeiten.
Entsprechend ist die Erforschung der molekularen Grundlagen dieser Erkrankungen von entscheidender Bedeutung. Sie kann dazu beitragen, präventive Maßnahmen zu entwickeln und auch Wege zur Wiederverwendung bereits zugelassener Medikamente für diese seltenen Fälle zu ebnen. Die Zusammenarbeit zwischen Wissenschaftlern und Klinikern ist daher unerlässlich für ein besseres Verständnis dieser seit langem vernachlässigten Erkrankungen. Am Freiburger Max-Planck-Institut für Immunbiologie und Epigenetik ist es nun in genau einer solchen Zusammenarbeit mit Klinikern aus Dijon gelungen, ein bisher unbekanntes und seltenes Syndrom aufzuklären. Bei diesem leiden betroffene Kinder an schweren Entwicklungsverzögerungen sowie fortschreitenden neurologischen Fehlfunktionen.
Mutation im MSL3-Gen
Die Zusammenarbeit begann, als Asifa Akhtar, Direktorin am Freiburger Max-Planck-Institut und führende Expertin für Epigenetik, von dem Kliniker Julien Thevenon von der Abteilung für Medizinische Genetik der Universität Dijon in Frankreich kontaktiert wurde. Mithilfe modernster Sequenzierungstechnologien zur Genomanalyse hatte er festgestellt, dass der Grund für die neurologische Entwicklungsstörung bei einem seiner jugendlichen Patienten durch die Mutation eines Gens namens MSL3 verursacht wurde. Um die konkreten Auswirkungen einer solchen Mutation auf den Menschen besser zu verstehen, fragte er die Unterstützung und Expertise der Freiburger Forscher an. „Zu diesem Zeitpunkt ist es aber auch wichtig, weitere Patienten mit ähnlichem Krankheitsbild und Mutationen im gleichen Gen zu finden, um die Diagnose zu bestätigen“, sagt Julien Thevenon, Co-Autor der Studie.
Durch das Matchmaker Exchange-Projekt, eine Plattform, die Kliniker und Forscher mit Diagnosen und Daten von seltenen Erkrankungen aus der ganzen Welt verbindet, konnte Thevenon fünfzehn weitere Patienten in Großbritannien, Frankreich, Deutschland, den Niederlanden, Dänemark, Estland, Belgien, den USA und Australien ausfindig machen. Alle von ihnen zeigten ähnliche Symptome und auch Veränderungen des MSL3-Gens. „Seit mehreren Jahren untersuchen wir das MSL3-Gen in verschiedenen Modellorganismen. Das war eine ganz wichtige Voraussetzung, um nun mit dieser schwierigen Aufgabe der Aufklärung zu beginnen, warum und wie der Verlust der MSL3-Funktion zu der Erkrankung beiträgt“, sagt Asifa Akhtar.
Das Forscherteam wusste bereits, dass die von den MSL-Genen kodierten Proteine, so z.B. MSL3, im Prinzip wie eine Art Lautstärkeregler eines Radios wirken: Sie sind in der Lage, ein Gen präzise auf das benötigte Niveau zu aktivieren. In langjährigen Studien zeigte das Labor von Asifa Akhtar, wie sich verschiedende MSL-Proteine zu dem sogenannten MSL-Komplex zusammenschließen, der beispielsweise das gesamte X-Chromosom von Fruchtfliegen regulieren kann aber auch einzelne Gene bei Säugetieren feinabstimmt. „Das komplexe Krankheitsbild der Patienten ließ uns vermuten, dass MSL3 während der Entwicklung verschiedene Rollen spielen könnte", sagt Felicia Basilicata, Hauptautorin der Studie.
Beeinträchtigte epigenetische Feinabstimmung von Entwicklungsgenen
Im Verlauf der Untersuchung in Freiburg konnten die Wissenschaftler zeigen, dass diese Feinabstimmung beim Menschen für bestimmte Entwicklungsgene besonders wichtig ist. Während der Embryonalentwicklung eines Kindes müssen Entwicklungsgene wie Zahnräder in einem Schweizer Uhrwerk funktionen. Ein präzises Zusammenwirken aller ist notwendig, um einen optimalen Verlauf der Entwicklung zu gewährleisten. Bei MSL3-Syndrom-Patienten sind die Entwicklungsgene selbst intakt, aber das epigenetische Programm reguliert durch den MSL-Komplex, das diese Feinabstimmung der genetischen Informationen reguliert, ist beeinträchtigt. Dies kann zur globalen Verzögerung der Entwicklung mehrerer Organe, einschließlich des Gehirns, führen.
Die Wissenschaftler hatten das Glück, dass einige der Patienten damit einverstanden waren, Hautbiopsien für die molekulare Forschung in Freiburg zu spenden. Dadurch war es dem Team möglich, histologische Studien durchzuführen und Zellkulturmodelle zu erstellen. So gelang es, die molekularen Mechanismen zu verstehen, die das Syndrom verursacht hatten. Die Wissenschaftler fanden heraus, dass das MSL3-Protein als Teil des MSL-Komplexes essentiell ist, um die Aktivität des Enzyms MOF zu steigern. Dieses Enzym, das ebenfalls Teil des MSL-Komplexes ist, markiert bestimmte Gene und macht sie dadurch leichter zugänglich, damit sie abgelesen werden können. „Die Mutationen, die wir im MSL3-Gen der Patienten beobachten konnten, führen zu einer Fehlregulation der enzymatischen Aktivität des MSL-Komplexes. Die beeinträchtigte Funktion dieses epigenetischen Regulators führt wiederum zu einer verminderten Anzahl epigenetischen Markierungen. Daraus resultiert eine reduzierte Aktivität zahlreicher, biologisch relevanter Gene, einschließlich wichtiger Entwicklungsregulatoren, die so zum Krankheitszustand der Patienten mit MSL3-Syndrom beitragen,“ fasst Asifa Akhtar die Folgen des mutierten Gens zusammen.
Epigenetische Therapie zur Wiederherstellung der Zellfunktion
Aber die Ergebnisse geben auch Anlass zur Hoffnung: Genetische Veränderungen verändern das Genom dauerhaft. Im Gegensatz dazu können epigenetische Veränderungen, unabhängig davon, ob sie durch Genomveränderungen, wie beispielsweise beim MSL3-Syndrom, oder durch Umwelteinflüsse wie Stress, Alterung oder Ernährung hervorgerufen werden, pharmakologisch rückgängig gemacht oder zumindest abgemildert werden. So entstand die Idee, die kompromittierte Funktion des MSL-Komplexes bei Patienten mit diagnostiziertem MSL3-Syndrom zu umgehen.
Die Freiburger Forscher hatten die Idee, Substanzen zu testen, die bekanntermaßen die Acetylierungsmarkierungen in den Zellen erhöhen. Diese Medikamente wurden ursprünglich für die Krebstherapie entwickelt und kürzlich in ersten klinischen Studien erfolgreich getestet. „Unsere Ergebnisse waren erstaunlich. Wir haben sogenannte Histondeacetylase-Inhibitoren in die MSL3-Syndromzellen appliziert und eine klare Verbesserung der behandelten Zellen festgestellt – sowohl auf molekularer als auch auf zellulärer Ebene", sagt Felicia Basilicata.
Obwohl der Therapieansatz zunächst nur in der Zellkultur getestet wurde, bietet er einen vorläufigen, aber vielversprechenden Startpunkt für die weitere Erforschung neuer Behandlungsmöglichkeiten. „Wir hoffen, dass wir durch die Identifizierung der Ursache und der zugrundeliegenden molekularen Mechanismen der Krankheit perspektivisch einige der Symptome von MSL3-Patienten lindern können. Und wir wünschen uns natürlich auch, dass unsrere Ergebnisse den Weg für weitere Studien ebnen, die zum Ziel haben Patienten mit MSL3-Syndrom zu helfen sowie dazu beitragen Krankheiten zu erforschen, bei denen ebenfalls epigenetische Regulatoren betroffen sind“, sagt Julien Thevenon. „Unsere Studie ist ein gutes Beispiel dafür, wie in einem integrativer Ansatz von Molekularwissenschaftlern und Klinikern Hand in Hand zusammengearbeitet wird, um die grundlegenden Mechanismen zu entschlüsseln, die zum Verständnis der zum Teil komplexen Ursachen menschlicher Krankheiten erforderlich sind“, sagt Asifa Akhtar.