Max-Planck-Institut für Immunbiologie und Epigenetik
Die Aufklärung des genregulatorischen Kodes im menschlichen Erbgut mittels quantitativer Proteomik
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für Immunbiologie und Epigenetik
Autoren
Mittler, Gerhard
Abteilungen
Zelluläre und molekulare Immunologie (Prof. Dr. Rudolf Grosschedl) MPI für Immunbiologie, Freiburg
Zusammenfassung
Das menschliche Genom besitzt etwa 25.000 Gene, welche die Bauanleitung für eine ebenso große Anzahl an Proteinen darstellen. In einem bestimmten Zelltyp werden aber nur maximal 10.000 dieser Gene in signifikanter Weise abgelesen. Um dies zu erklären, ist es unabdingbar, den genregulatorischen Kode zu verstehen, der durch eine besondere Klasse von DNS-bindenden Proteinen, den Transkriptionsfaktoren (TFs), ausgelesen wird. Forscher am MPI für Immunbiologie haben eine schnelle und hochempfindliche Methodik entwickelt, die es ermöglichen könnte, den genregulatorischen Kode des ganzen menschlichen Genoms zu entziffern.
Thematische Einführung
Die differentielle Gewebe-spezifische Genexpression stellt einen wichtigen Bestandteil der Genregulation beim Menschen dar. Darüber hinaus ist die Expression bestimmter Gene ausschließlich auf bestimmte Entwicklungsstadien der Embryonalentwicklung beschränkt, und das fälschliche Ablesen von Genen geht oft mit der Ausbildung von Tumoren sowie der Entwicklung anderer Krankheiten einher. Deshalb ist ein detailliertes Verständnis der zeitlichen und räumlichen Genexpressionsmuster von grundlegender Bedeutung für das Verständnis der Komplexität des Humangenoms. Dies stellt keine leichte Aufgabe dar, da die Wörter dieser Sprache (cis-Elemente genannt) meist mehrdeutig und Kontext-abhängig fungieren und sowohl in der unmittelbaren Nähe von Genen als auch tausende von DNS-Basenpaaren entfernt angetroffen werden können.
Eine elegante Studie, die kürzlich in der angesehenen Fachzeitschrift Science publiziert wurde, konnte in eindrucksvoller Weise zeigen, dass das Genexpressionsmuster des menschlichen Chromosoms 21 – eingebracht in die Maus – dort identisch zum Expressionsmuster in menschlichen Zellen ist [1]. Dies macht deutlich, dass regulatorische cis-Elemente und nicht andere Spezies-spezifische Faktoren (z.B. epigenetische Effekte) primär für das Genexpressionsprogramm von Chromosom 21 verantwortlich sind. Damit rückt die Aufklärung des genregulatorischen Kodes wieder in den Brennpunkt des allgemeinen wissenschaftlichen Interesses [2]. Die vor uns liegende Aufgabe lässt sich somit in erster Näherung auf eine simple Fragestellung reduzieren: „Welcher oder welche Transkriptionsfaktoren (TFs) binden in einem gegebenen Zelltyp spezifisch an ein regulatorisches cis-Element?“ Die experimentelle Beantwortung dieser Frage wird durch zwei Faktoren wesentlich erschwert. Zum einen stellt die DNS-Doppelhelix physikochemisch ein Polyanion dar, welches Sequenz-unabhängige ionische Wechselwirkungen mit stark positiv geladenen Proteinen eingehen kann. Diesen Umstand macht sich die Natur sogar in allen eukaryontischen Zellen bei der Verpackung der Erbsubstanz zu Nutze: Mittels der stark positiv geladenen Histonproteine entstehen so genannte Nukleosomen, welche die kleinste Organisationseinheit eines Chromosoms darstellen. Zum anderen sind Transkriptionsfaktoren vergleichsweise seltene Proteine, sodass deren Nachweis empfindliche analytische Methoden erfordert [3].
Traditionelle Methoden zur Bestimmung von DNS-Protein-Interaktionen
Bis vor kurzem konnten Forscher nur über einen zeitaufwändigen, kombinierten experimentellen Ansatz die Bindung eines Transkriptionsfaktors an ein cis-Element aufklären [4]. Mithilfe eines so genannten Gelretardations-Assays (EMSA: Electrophoretic Mobility Shift Assay) können spezifische DNS-TF-Komplexe von unspezifischen DNS-Proteinkomplexen abgetrennt und auf einem Polyacrylamidgel sichtbar gemacht werden (Abb. 1). Als Ausgangsmaterial für diese Experimente dienen in der Regel Proteinextrakte aus den Zellkernen menschlicher Zelllinien. In einem nächsten Schritt muss nun der Transkriptionsfaktor aus dem verwendeten Proteingemisch mittels biochemischer Methoden isoliert und zur Homogenität gereinigt werden. Dies erfordert meist mehrere Chromatographie-Reinigungsschritte, wobei in der Regel im letzten Schritt die DNA-Bindestelle des gesuchten TFs als Ligand in einer so genannten Affinitätschromatographie eingesetzt wird. Die erfolgreiche Anreicherung des gesuchten Transkriptionsfaktors in diesem mehrstufigen Prozess wird dabei wiederum mithilfe des Gelretardations-Assays dokumentiert [4]. Schließlich ermittelt man die Identität des gereinigten TFs (durch seine Aminosäuresequenz bestimmt) durch massenspektrometrische Verfahren, welche im nächsten Abschnitt näher erläutert werden.
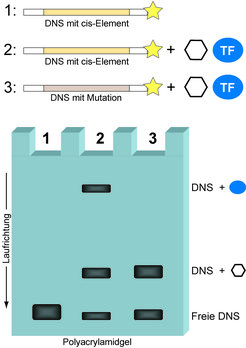
Der Gelretardations-Assay (engl. EMSA, siehe Text) zeigt das Vorhandensein eines Transkriptionsfaktors (TF) auf, der einen gegebenen regulatorischen DNS-Abschnitt (cis-Element) Sequenz-spezifisch binden kann. Radioaktiv markierte (gelber Stern) DNS, welche für ein cis-Element kodiert, wird entweder ohne (1) oder in Anwesenheit (2) eines Proteinextrakts aus humanen Zellen in einem Reagenzgefäß unter nahezu physiologischen Bedingungen inkubiert. Als Kontrolle dient dabei der entsprechende DNS-Abschnitt in seiner mutierten Form (3). Dabei kommt es zur Ausbildung von DNS-Proteinkomplexen, was wiederum in einem nativen Polyacrylamidgel zu einer Retardation der DNS-Moleküle im elektrischen Feld führt (Gelspuren 2 und 3). Nur der Sequenz-spezifisch interagierende TF generiert eine charakteristische retardierte Gelbande (Spur 2), während unspezifisch bindende Proteine (als Sechseck dargestellt) sowohl die natürliche als auch die mutierte DNS schwach retardieren (Spuren 2 und 3). Die Aussage des Experiments ist allerdings limitiert, da die molekulare Identität des TFs unbekannt bleibt (siehe Text). Deshalb wurde der SILAC DNS-Proteininteraktions-Screen (Abb. 2) entwickelt.
Der Gelretardations-Assay (engl. EMSA, siehe Text) zeigt das Vorhandensein eines Transkriptionsfaktors (TF) auf, der einen gegebenen regulatorischen DNS-Abschnitt (cis-Element) Sequenz-spezifisch binden kann. Radioaktiv markierte (gelber Stern) DNS, welche für ein cis-Element kodiert, wird entweder ohne (1) oder in Anwesenheit (2) eines Proteinextrakts aus humanen Zellen in einem Reagenzgefäß unter nahezu physiologischen Bedingungen inkubiert. Als Kontrolle dient dabei der entsprechende DNS-Abschnitt in seiner mutierten Form (3). Dabei kommt es zur Ausbildung von DNS-Proteinkomplexen, was wiederum in einem nativen Polyacrylamidgel zu einer Retardation der DNS-Moleküle im elektrischen Feld führt (Gelspuren 2 und 3). Nur der Sequenz-spezifisch interagierende TF generiert eine charakteristische retardierte Gelbande (Spur 2), während unspezifisch bindende Proteine (als Sechseck dargestellt) sowohl die natürliche als auch die mutierte DNS schwach retardieren (Spuren 2 und 3). Die Aussage des Experiments ist allerdings limitiert, da die molekulare Identität des TFs unbekannt bleibt (siehe Text). Deshalb wurde der SILAC DNS-Proteininteraktions-Screen (Abb. 2) entwickelt.
Die quantitative Peptid-Massenspektrometrie identifiziert Sequenz-spezifisch DNS-bindende Transkriptionsfaktoren
Um den traditionellen Ansatz entscheidend zu verbessern, ist es unabdingbar, die Isolierung und Anreicherung des Transkriptionsfaktors direkt mit dem Nachweis seiner Sequenzspezifität sowie seiner Identifikation zu koppeln. Man macht sich dabei den Umstand zu Nutze, dass artifizielle Veränderungen/Mutationen in der DNS-Sequenz (Austausch von Basenpaaren) eines cis-Elements dessen Funktion zerstören und somit die Bindung des TFs verhindert wird. Dieses Verhalten lässt sich experimentell durch die bereits erwähnte DNS-Affinitätschromatographie studieren, bei der sowohl die natürliche als auch die mutierte Sequenz (Kontrolle) in Form eines an die Säulenmatrix immobilisierten Liganden zum Einsatz kommt. Ein direkter quantitativer Vergleich des Proteinbindungsmusters zwischen der natürlichen und der mutierten DNS-Sequenz liefert somit die Identität des Transkriptionsfaktors, welcher für die Funktion des regulatorischen Elements verantwortlich ist. Beim SILAC DNS-Proteininteraktions-Screen erfolgt die differentielle Bindungsmusteranalyse direkt in einem modernen hochauflösenden Massenspektrometer.
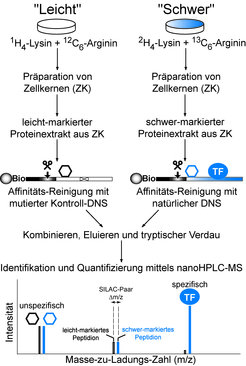
Das experimentelle Vorgehen soll nun im Folgenden kurz skizziert werden. Nach der DNS-Affinitätschromatographie werden sowohl die Proteine, die an die natürliche DNS-Sequenz, als auch diejenigen Polypeptide, welche an die mutierte Kontroll-DNS binden, durch die Endoproteinase Trypsin verdaut (Trypsin hydrolysiert die Peptidbindung sequenzspezifisch nach Arginin- oder Lysinresten): Es entsteht ein hochkomplexes Peptidgemisch. Dieses wird durch die Kopplung einer Hochdruckflüssigkeitschromatographie-Anlage mit dem Massenspektrometer aufgetrennt, ionisiert und in der Gasphase analysiert. Die Signale/Peaks in den Massenspektren liefern im Wesentlichen drei Informationen: (1) Die Molekülmasse der Peptide (berechnet aus dem gemessenen Masse-zu-Ladungs-Verhältnis), die (2) Molekülmasse von Fragment-Ionen der Peptide (liefert zusammen mit (1) letztlich die Peptidaminosäuresequenz) und die (3) Intensitäten der Peaks, welche für die anschließende Quantifizierung verwendet werden [5]. Für die Quantifizierung wird zudem ein weiterer Parameter wichtig, der bisher nicht diskutiert wurde. Tryptische Peptide der Proteine, die an die natürliche DNS gebunden waren, können im Massenspektrum anhand einer charakteristischen Verschiebung des Molekulargewichts zu höheren Massen, welche durch nicht-radioaktive Isotopenmarkierung erreicht wird, direkt erkannt werden. Die Isotopenmarkierung erfolgt dabei durch metabolischen Einbau von „schwerem“ Deuterium-markiertem Lysin (2H4-Lysin) und „schwerem“ 13-Kohlenstoff-markiertem Arginin (13C6-Arginin) bei der Gewebekultur menschlicher Zelllinien. Als Referenzkultur für das Kontrollexperiment dient eine Gewebekultur mit den natürlich vorkommenden „leichten“ (unmarkierten) Aminosäuren. Diese Methodik wurde von Matthias Mann und Kollegen (Max-Planck-Institut für Biochemie) entwickelt und als SILAC (Stable Isotope Labeling of Amino acids in Cell culture) bezeichnet [6]. Der große Vorteil dabei ist, dass man die relativen Peptidmengen der Proteine, welche an das cis-Element bzw. die Kontroll-DNS binden, direkt im gleichen Massenspektrum, sprich in der gleichen Messung bestimmen kann, was eine äußerst genaue Quantifizierung ermöglicht. Wie in Abb. 2 gezeigt, weist der spezifisch-bindende Transkriptionsfaktor also nur „schwere“ isotopenmarkierte Peptidionensignale auf, da das „leicht“-markierte Protein kaum mit der mutierten DNS (Kontrollsäule) interagieren kann.
Der SILAC DNS-Proteininteraktions-Screen kombiniert den Nachweis der Sequenzspezifität eines Transkriptionsfaktors mit der Aufklärung seiner molekularen Identität (Aminosäuresequenz). Die SILAC-Methode (Stable Isotope Labeling of Amino acids in Cell culture) ermöglicht die differentielle metabolische Markierung des zellulären Proteoms durch den alternativen Einbau von natürlichen leicht-markierten bzw. chemisch-synthetisierten schwer-markierten Aminosäuren. Der Einsatz von Lysin und Arginin garantiert im Zusammenspiel mit dem tryptischen Verdau der Probe (tryptische Peptide tragen entweder einen Lysin- oder einen Argininrest an ihrem C-Terminus) eine komplette Markierung der Proteome. Zur Anreicherung DNS-bindender Proteine wird eine einstufige DNS-Affinitätsreinigung durchgeführt. Die Affinitätssäule besteht aus biotinylierter (Bio) DNS, welche an einer Streptavidin-Matrix (interagiert äußerst stark mit Biotin) immobilisiert wurde. Die Elution der DNS-Proteinkomplexe erfolgt schonend mit Hilfe eines DNS-schneidenden Restriktionsenzyms (als Schere illustriert). Da der TF praktisch nicht an die mutierte Kontroll-DNS-Affinitätsmatrix bindet, liefert er nach tryptischem Verdau und nanoHPLC-MS-Analyse nahezu ausschließlich schwer-markierte Peptidionen im Massenspektrum. Im Gegensatz dazu entstehen aus unspezifisch DNS-bindenden Proteinen (als Sechseck dargestellt), welche den Hauptanteil (99%) der Probe ausmachen, leicht- bzw. schwer-markierte Peptidionen gleicher Intensität. Leicht-markierte und schwer-markierte Peptidionen, die eine identische Aminosäuresequenz aufweisen, werden im Massenspektrum als sogenannte SILAC-Paare beobachtet. Letztere zeichnen sich durch eine charakteristische Massenverschiebung (Δm/z) in ihrem Molekulargewicht aus.
Der SILAC DNS-Proteininteraktions-Screen kombiniert den Nachweis der Sequenzspezifität eines Transkriptionsfaktors mit der Aufklärung seiner molekularen Identität (Aminosäuresequenz). Die SILAC-Methode (Stable Isotope Labeling of Amino acids in Cell culture) ermöglicht die differentielle metabolische Markierung des zellulären Proteoms durch den alternativen Einbau von natürlichen leicht-markierten bzw. chemisch-synthetisierten schwer-markierten Aminosäuren. Der Einsatz von Lysin und Arginin garantiert im Zusammenspiel mit dem tryptischen Verdau der Probe (tryptische Peptide tragen entweder einen Lysin- oder einen Argininrest an ihrem C-Terminus) eine komplette Markierung der Proteome. Zur Anreicherung DNS-bindender Proteine wird eine einstufige DNS-Affinitätsreinigung durchgeführt. Die Affinitätssäule besteht aus biotinylierter (Bio) DNS, welche an einer Streptavidin-Matrix (interagiert äußerst stark mit Biotin) immobilisiert wurde. Die Elution der DNS-Proteinkomplexe erfolgt schonend mit Hilfe eines DNS-schneidenden Restriktionsenzyms (als Schere illustriert). Da der TF praktisch nicht an die mutierte Kontroll-DNS-Affinitätsmatrix bindet, liefert er nach tryptischem Verdau und nanoHPLC-MS-Analyse nahezu ausschließlich schwer-markierte Peptidionen im Massenspektrum. Im Gegensatz dazu entstehen aus unspezifisch DNS-bindenden Proteinen (als Sechseck dargestellt), welche den Hauptanteil (99%) der Probe ausmachen, leicht- bzw. schwer-markierte Peptidionen gleicher Intensität. Leicht-markierte und schwer-markierte Peptidionen, die eine identische Aminosäuresequenz aufweisen, werden im Massenspektrum als sogenannte SILAC-Paare beobachtet. Letztere zeichnen sich durch eine charakteristische Massenverschiebung (Δm/z) in ihrem Molekulargewicht aus.
Charakterisierung eines hochkonservierten Promoterelements in menschlichen Zellen
Das Prinzip des SILAC DNS-Proteininteraktions-Screens wurde kürzlich von Max-Planck-Wissenschaftlern beschrieben [7]. Die Technologie wird nun am MPI für Immunologie routinemäßig von Forschern eingesetzt – ein aktuelles Anwendungsgebiet soll hier kurz skizziert werden. Die Promoterregionen menschlicher Gene befinden sich in unmittelbarer Nähe des Transkriptionsstartes, sprich derjenigen Stelle auf der DNS, an der die Ablesung eines Gens beginnen kann. Es ist bekannt, dass Promoterregionen eine hoche Dichte genregulatorischer cis-Elemente aufweisen, von denen eine Vielzahl in den letzten dreißig Jahren untersucht wurde. Moderne bioinformatische Analysen der Promoterregionen haben nun allerdings viele in Säugern evolutionär hochkonservierte cis-Elemente entdeckt, deren Funktion bisher unbekannt war [8;9]. Das am besten konservierte Element (bezeichnet als HCSM1, Hematopoietic Cell Specific Motif 1) mit der DNS-Konsensussequenz "RRACTACANNTCCCRRNRNRC" wird (wahrscheinlich zumindest teilweise gleichzeitig) von mehreren Transkriptionsfaktoren gebunden, welche man drei TF-Familien zuordnen kann: der Ikaros-, der NFkB/Rel- und der THAP-Familie. Die Bindung dieser Proteine an die HCSM1-Gene in lebenden Zellen wurde zudem in unabhängigen Experimenten mittels Chromatin Immuno-Precipitation bestätigt (zur Methodik siehe [10]). Dies zeigt deutlich, dass die von Max-Planck-Forschern entwickelte SILAC DNS-Proteininteraktions-Screening-Technologie einen entscheidenden Beitrag zum Verständnis des genregulatorischen Kodes leisten kann und leisten wird. Aufgrund der prominenten Rolle der Ikaros- beziehungsweise NFkB/Rel-TFs in blutbildenden (hämatopoietischen) Zellen haben die Forscher am MPI für Immunbiologie nun damit begonnen, die funktionelle Rolle dieser Proteine für die Regulation der HCSM1-Gene in hämatopoietischen Zellen zu studieren. Über die Proteine der THAP-Familie ist bis dato nicht sehr viel bekannt, es gibt aber Hinweise, dass sie an der Steuerung von Genen beteiligt sind, deren Regulation wichtig für die Stammzelleigenschaften bestimmter Zellen sind. Deshalb soll zukünftig auch eine mögliche Funktion des HCSM1-Elements im Kontext blutbildender Stammzellen untersucht werden.
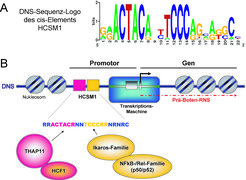
Die DNS-Sequenz des HCSM1 cis-Elements wird in vivo durch mehrere Transkriptionsfaktoren ausgelesen.
(A) Durch Sequenzvergleich der HCSM1-Elemente, welche in mehr als 500 menschlichen Genpromotoren gefunden werden können, lässt sich ein so genanntes DNS-Sequenz-Logo erstellen. Dieses zeigt die relative Häufigkeit einer Base an einer bestimmten Position in der DNS-Sequenz auf. Die Analyse macht deutlich, dass das HCSM1-Motiv zwei stark konservierte invariante Abschnitte mit der Sequenz ACTACA bzw. TCCC aufweist. Invariante Basen sind in der Regel wichtige Bindungsstellen für Sequenz-spezifische TFs.
(B) Schematische Darstellung der Organisation eines Gens mit einem HCSM1-Element in seiner Promotorregion. Die DNS liegt in ihrer natürlichen Form als Chromatin in Nukleosomen (grau gefüllte Kreise) verpackt vor. Die Transkriptionsmaschine erledigt, reguliert durch TFs, die Synthese der Vorläufer-Boten-RNS, welche später als Blaupause für die Proteinbiosynthese an den Ribosomen dient. Die Transkriptionsstartstelle ist durch einen Pfeil gekennzeichnet. Das HCSM1-Motiv befindet sich stromaufwärts in unmittelbarer Nähe des Transkriptionsstartpunkts. Der aktuelle Wissensstand lässt sich wie folgt zusammenfassen: Das THAP11-Protein sowie die TFs Ikaros, Helios und Aiolos (Ikaros-Proteinfamilie) sind in der Lage, voneinander unabhängig die beiden invarianten DNS-Sequenzabschnitte (ACTACR bzw. TCCCRR) auszulesen. HCF-1 sowie die Proteine p50 und p52 (aus der NFkB-/Rel-Familie) werden vorwiegend durch Protein-Protein-Interaktionen mit THAP11 bzw. Ikaros an das cis-Element rekrutiert.
THAP (Thanatos associated protein), HCF-1 (Host Cell Factor-1), NFkB (Nuclear Factor kappaB), HCSM1 (Hematopoietic Cell Specific Motif 1).
Die DNS-Sequenz des HCSM1 cis-Elements wird in vivo durch mehrere Transkriptionsfaktoren ausgelesen.
(A) Durch Sequenzvergleich der HCSM1-Elemente, welche in mehr als 500 menschlichen Genpromotoren gefunden werden können, lässt sich ein so genanntes DNS-Sequenz-Logo erstellen. Dieses zeigt die relative Häufigkeit einer Base an einer bestimmten Position in der DNS-Sequenz auf. Die Analyse macht deutlich, dass das HCSM1-Motiv zwei stark konservierte invariante Abschnitte mit der Sequenz ACTACA bzw. TCCC aufweist. Invariante Basen sind in der Regel wichtige Bindungsstellen für Sequenz-spezifische TFs.
(B) Schematische Darstellung der Organisation eines Gens mit einem HCSM1-Element in seiner Promotorregion. Die DNS liegt in ihrer natürlichen Form als Chromatin in Nukleosomen (grau gefüllte Kreise) verpackt vor. Die Transkriptionsmaschine erledigt, reguliert durch TFs, die Synthese der Vorläufer-Boten-RNS, welche später als Blaupause für die Proteinbiosynthese an den Ribosomen dient. Die Transkriptionsstartstelle ist durch einen Pfeil gekennzeichnet. Das HCSM1-Motiv befindet sich stromaufwärts in unmittelbarer Nähe des Transkriptionsstartpunkts. Der aktuelle Wissensstand lässt sich wie folgt zusammenfassen: Das THAP11-Protein sowie die TFs Ikaros, Helios und Aiolos (Ikaros-Proteinfamilie) sind in der Lage, voneinander unabhängig die beiden invarianten DNS-Sequenzabschnitte (ACTACR bzw. TCCCRR) auszulesen. HCF-1 sowie die Proteine p50 und p52 (aus der NFkB-/Rel-Familie) werden vorwiegend durch Protein-Protein-Interaktionen mit THAP11 bzw. Ikaros an das cis-Element rekrutiert.
THAP (Thanatos associated protein), HCF-1 (Host Cell Factor-1), NFkB (Nuclear Factor kappaB), HCSM1 (Hematopoietic Cell Specific Motif 1).
 zeigt das Vorhandensein eines Transkriptionsfaktors (TF) auf, der einen gegebenen regulatorischen DNS-Abschnitt (cis-Element) Sequenz-spezifisch binden kann. Radioaktiv markierte (gelber Stern) DNS, welche für ein cis-Element kodiert, wird entweder ohne (1) oder in Anwesenheit (2) eines Proteinextrakts aus humanen Zellen in einem Reagenzgefäß unter nahezu physiologischen Bedingungen inkubiert. Als Kontrolle dient dabei der entsprechende DNS-Abschnitt in seiner mutierten Form (3). Dabei kommt es zur Ausbildung von DNS-Proteinkomplexen, was wiederum in einem nativen Polyacrylamidgel zu einer Retardation der DNS-Moleküle im elektrischen Feld führt (Gelspuren 2 und 3). Nur der Sequenz-spezifisch interagierende TF generiert eine charakteristische retardierte Gelbande (Spur 2), während unspezifisch bindende Proteine (als Sechseck dargestellt) sowohl die natürliche als auch die mutierte DNS schwach retardieren (Spuren 2 und 3). Die Aussage des Experiments ist allerdings limitiert, da die molekulare Identität des TFs unbekannt bleibt (siehe Text). Deshalb wurde der SILAC DNS-Proteininteraktions-Screen (Abb. 2) entwickelt.")
. Die SILAC-Methode (Stable Isotope Labeling of Amino acids in Cell culture) ermöglicht die differentielle metabolische Markierung des zellulären Proteoms durch den alternativen Einbau von natürlichen leicht-markierten bzw. chemisch-synthetisierten schwer-markierten Aminosäuren. Der Einsatz von Lysin und Arginin garantiert im Zusammenspiel mit dem tryptischen Verdau der Probe (tryptische Peptide tragen entweder einen Lysin- oder einen Argininrest an ihrem C-Terminus) eine komplette Markierung der Proteome. Zur Anreicherung DNS-bindender Proteine wird eine einstufige DNS-Affinitätsreinigung durchgeführt. Die Affinitätssäule besteht aus biotinylierter (Bio) DNS, welche an einer Streptavidin-Matrix (interagiert äußerst stark mit Biotin) immobilisiert wurde. Die Elution der DNS-Proteinkomplexe erfolgt schonend mit Hilfe eines DNS-schneidenden Restriktionsenzyms (als Schere illustriert). Da der TF praktisch nicht an die mutierte Kontroll-DNS-Affinitätsmatrix bindet, liefert er nach tryptischem Verdau und nanoHPLC-MS-Analyse nahezu ausschließlich schwer-markierte Peptidionen im Massenspektrum. Im Gegensatz dazu entstehen aus unspezifisch DNS-bindenden Proteinen (als Sechseck dargestellt), welche den Hauptanteil (99%) der Probe ausmachen, leicht- bzw. schwer-markierte Peptidionen gleicher Intensität. Leicht-markierte und schwer-markierte Peptidionen, die eine identische Aminosäuresequenz aufweisen, werden im Massenspektrum als sogenannte SILAC-Paare beobachtet. Letztere zeichnen sich durch eine charakteristische Massenverschiebung (Δm/z) in ihrem Molekulargewicht aus.")
 Durch Sequenzvergleich der HCSM1-Elemente, welche in mehr als 500 menschlichen Genpromotoren gefunden werden können, lässt sich ein so genanntes DNS-Sequenz-Logo erstellen. Dieses zeigt die relative Häufigkeit einer Base an einer bestimmten Position in der DNS-Sequenz auf. Die Analyse macht deutlich, dass das HCSM1-Motiv zwei stark konservierte invariante Abschnitte mit der Sequenz ACTACA bzw. TCCC aufweist. Invariante Basen sind in der Regel wichtige Bindungsstellen für Sequenz-spezifische TFs.
(B) Schematische Darstellung der Organisation eines Gens mit einem HCSM1-Element in seiner Promotorregion. Die DNS liegt in ihrer natürlichen Form als Chromatin in Nukleosomen (grau gefüllte Kreise) verpackt vor. Die Transkriptionsmaschine erledigt, reguliert durch TFs, die Synthese der Vorläufer-Boten-RNS, welche später als Blaupause für die Proteinbiosynthese an den Ribosomen dient. Die Transkriptionsstartstelle ist durch einen Pfeil gekennzeichnet. Das HCSM1-Motiv befindet sich stromaufwärts in unmittelbarer Nähe des Transkriptionsstartpunkts. Der aktuelle Wissensstand lässt sich wie folgt zusammenfassen: Das THAP11-Protein sowie die TFs Ikaros, Helios und Aiolos (Ikaros-Proteinfamilie) sind in der Lage, voneinander unabhängig die beiden invarianten DNS-Sequenzabschnitte (ACTACR bzw. TCCCRR) auszulesen. HCF-1 sowie die Proteine p50 und p52 (aus der NFkB-/Rel-Familie) werden vorwiegend durch Protein-Protein-Interaktionen mit THAP11 bzw. Ikaros an das cis-Element rekrutiert.
THAP (Thanatos associated protein), HCF-1 (Host Cell Factor-1), NFkB (Nuclear Factor kappaB), HCSM1 (Hematopoietic Cell Specific Motif 1).")