Das Adapterprotein SLP-65 als Tumorsuppressor bei der häufigsten Leukämie im Kindesalter
Forschungsbericht (importiert) 2003 - Max-Planck-Institut für Immunbiologie und Epigenetik
Leukämien sind tumorartige Erkrankungen des Knochenmarks, bei denen unreife Vorläuferzellen der weißen Blutkörperchen entarten und sich dann ungehemmt vermehren. Bei etwa 80 Prozent der Leukämien im Kindes- und Jugendalter handelt es sich um die akute lymphatische Leukämie (ALL), die somit die häufigste Leukämie bei Kindern ist. Diese Tumoren entstehen aus Prä-B-Zellen, den Vorläuferzellen der B-Lymphozyten. Die B-Zellen sind ein wichtiger Teil unseres humoralen Immunsystems. Nach ihrer Differenzierung zu Plasmazellen produzieren diese Zellen die Antikörper, welche uns vor Infektionen schützen. Aus Stammzellen des Blutes entstehen ständig große Mengen von B-Zellen in unserem Körper. Die mit dieser Entstehung einhergehende starke Zellteilung macht die Prä-B-Zellen besonders anfällig für Entartungen, die zu Leukämien führen können. Bisher haben die Wissenschaftler angenommen, dass diese Leukämien vor allem durch chromosomale Veränderungen, die im Genom dieser Leukämiezellen öfters gefunden werden, entstehen. Dabei handelt es sich entweder um Chromosomenvermehrungen oder um Chromosomenbrüche, bei denen Fusionsgene entstehen können, die zur Herstellung von Proteinen mit tumorerzeugenden Eigenschaften führen. In den B-ALL-Tumoren sind die auftretenden Chromosomenbrüche jedoch nicht homogen und werden auch nicht in allen Fällen gefunden. Dies lässt vermuten, dass sie erst nach einer ersten Entartung der Prä-B-Zellen entstanden sind. Welche primären Ursachen zur Entstehung der akuten lymphatischen Leukämie aus Prä-B-Zellen führen, ist bisher nicht geklärt.
Ein wichtiger Hinweis zur möglichen Entstehung dieser Tumorerkrankung kommt, wie so oft, aus der Grundlagenforschung. Hier bearbeiten die Wissenschaftler zum Teil ganz andere Fragestellungen, die doch auf Umwegen zu neuen Erkenntnissen über Krankheiten des Menschen führen können. Das langjährige Forschungsinteresse der Abteilung für molekulare Immunologie besteht darin, genau zu verstehen, wie B-Zellen über ihren B-Zellantigenrezeptor (BCR) zur Zellteilung und Differenzierung angeregt werden. Eines der frühesten Ereignisse in diesem Stimulationsprozess ist die Aktivierung von Protein-Tyrosin-Kinasen (PTK), welche unterhalb des BCR eine ganze Reihe von PTK-Substratproteinen phosphorylieren. Mittels einer biochemischen Aufreinigung konnten die Wissenschaftler um Michael Reth eine Reihe dieser Substrate erstmalig identifizieren und näher charakterisieren. Hierbei haben sie unter anderem das Adapterprotein SLP-65 gefunden, welches nach einer Stimulation des BCR von der BCR-assoziierten Kinase Syk phosphoryliert wird und dann eine ganze Reihe anderer Signalproteine binden und organisieren kann (Abb. 1). Parallel zu den Arbeiten in Freiburg wurde dieses Adapterprotein ebenfalls von Andrew Chan und von Daisuke Kitamura in den USA und Japan gefunden und dort BLNK oder BASH genannt.
: Der BCR besteht aus antigenbindenden Domänen und den signalleitenden Untereinheiten Ig-a/Ig-b. Nach Antigenbindung wird die Proteintyrosinkinase Syk aktiviert und phosphoryliert SLP-65. Phosphoryliertes SLP-65 bietet Bindestellen für weitere essentielle Signalproteine wie Btk und PLC-γ, die dadurch zu aktivierten Rezeptoren rekrutiert werden. PLC-γ wird sowohl von Syk als auch Btk phosphoryiert und dadurch aktiviert. Aktivierte PLC-γ hydrolysiert das Membranlipid Phosphatidylinositol-Bis-Phosphat zu Inositol-Tris-Phosphat (IP3) und Diacylglycerol (DAG). IP3 und DAG sind wichtige Botenstoffe, die weitere Signalkaskaden in Gang setzen und die Differenzierung von Zellen bewirken.")

Adapterproteine können mehrere intrazelluläre Signalproteine zu einem größeren Proteinkomplex zusammenfassen und ermöglichen so die stringente Regulierung und schnelle Verarbeitung intrazellulärer Signale. Die Arbeiten mehrerer Gruppen zeigten, dass das Adapterprotein SLP-65 unter anderem das Enzym Phospholipase-Cγ (PLCγ) sowie die Bruton-Tyrosine-Kinase (BTK) bindet. Von diesen Bindungspartnern ist vor allem BTK recht bekannt, da Defekte in diesem Protein zu einer bekannten menschlichen Immundefizienzerkrankung, nämlich der "Bruton Disease" oder "X-linked Agammaglobulinemia (XLA)" führt.
Der überraschende Phänotyp SLP-65-defizienter Mäuse
Um die Bedeutung des Adapterproteins SLP-65 für die Entwicklung und Funktion der B-Zellen zu bestimmen, haben die Freiburger Wissenschaftler SLP-65-defiziente (knock-out) Mäuse hergestellt. Diese Mäuse zeigen eindeutig, dass das Adapterprotein SLP-65 für die normale B- Zellentwicklung wichtig ist. Ganz ähnlich wie Mäuse, denen das BTK-Gen fehlt, sind die SLP-65-defizienten Mäuse immundefizient. Sie besitzen kaum reife B-Zellen und produzieren weniger Antikörper. Überraschend war allerdings die Entdeckung, dass die Prä-B-Zellen aus diesen SLP-65-defizienten Mäusen eine verstärkte Zellteilungsaktivität aufweisen und dass einige dieser Mäuse Prä-B-Zelltumoren entwickeln. Zudem können SLP-65-defiziente Prä-B-Zellen sehr leicht in vitro mithilfe des Wachstumsfaktors IL-7 kultiviert werden. Wir konnten so eine Reihe von Prä-B-Zelllinien herstellen. Wurden diese Zelllinien in Mäuse zurückgegeben, so entwickelten sie ebenfalls Prä-B-Zelltumoren. Mittels retroviraler Vektoren wurde dann das SLP-65-Protein in den SLP-65- defizienten Prä-B-Zellen wieder zur Expression gebracht. Es zeigte sich dabei, dass SLP-65-positive Prä-B-Zellen nicht mehr in der Lage sind, nach Injektion in die Mäuse Tumoren zu entwickeln. Das Adapterprotein SLP-65 kann das Tumorwachstum direkt verhindern und ist somit ein Tumorsuppressor. In weiteren Untersuchungen zeigten die Immunbiologen, dass in Gegenwart von SLP-65 der Prä-B-Zellrezeptor internalisiert wird, das heißt nach Aktivierung in das Zellinnere zurücktransportiert wird, und die Zellen eine verstärkte Differenzierung durchführen. Zudem ist die Expression des Prä-B-Zellrezeptors für die verstärkte Zellteilung der Prä-B-Zellen notwendig.
Nach diesen Untersuchungen fragten sich die Forscher, ob das SLP-65-Gen möglicherweise auch eine Rolle bei menschlichen Prä-B-Zellleukämien spielen könnte. In Zusammenarbeit mit Kinderkliniken in Freiburg, Hannover, Zürich, Hamburg und Kuopio (Finnland) untersuchten sie eine Vielzahl von Prä-B-ALL-Proben junger Leukämiepatienten und stellten dabei tatsächlich fest, dass fast in der Hälfte dieser Prä-B-ALL- Tumoren das Adapterprotein SLP-65 fehlt oder in veränderter Form exprimiert wird.
Die alternative Prozessierung der SLP-65-mRNA
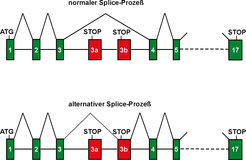
Wie kommt es zum Verlust des SLP-65-Adapterproteins, das ja normalerweise von allen B- Zellen ausgeprägt wird? Die B-ALL scheint keine vererbbare, sondern eine erworbene Krankheit zu sein. Die weiteren Untersuchungen zeigten dann auch, dass das menschliche SLP-65-Gen in den Tumorzellen der Patienten keinen Defekt aufwies. Vielmehr war die Ausprägung dieses Gens bei der Zwischenstufe der Boten- oder "messenger" RNA (mRNA) gestört. Bei den meisten menschlichen Genen liegt die Protein-kodierende Sequenz nicht in einem Stück vor, sondern ist in Sequenzblöcke unterteilt, die Exons genannt werden. Diese Exons sind auf der DNA durch mehr oder weniger große Intronsequenzen getrennt. Exonsequenzen werden erst auf der Ebene der mRNA zusammengefügt, in einem Prozess, der "Splicing" genannt wird. Genau bei diesem Prozess treten bei den SLP-65- defizienten Leukämiezellen Fehler auf. Das menschliche SLP-65-Gen besitzt zwischen Exon 3 und Exon 4 weitere alternative Exone (3a und 3b), welche bei dem normalen Splice-Prozess der SLP-65-mRNA nicht eingebaut werden (Abb. 2). Bei den SLP-65-defizienten B-ALL-Zellen werden jedoch diese alternativen Exons verstärkt in die mRNA eingebaut. Die alternativen SLP-65- Exons besitzen einen translationalen Stopp im Leseraster der SLP-65-mRNA. Somit kann nach ihrem Einbau kein vollständiges SLP-65-Protein mehr gebildet werden.
 unterteilt, die durch Introns (durchgezogene Linien) getrennt sind. Exons 6-16 und die dazwischen liegenden Introne sind als gestrichelte Linie vereinfacht dargestellt. Rote Vierecke symbolisieren alternative Exons, die keine kodierende SLP-65-Sequenz tragen. Das \"ATG\" ist das Startcodon für die SLP-65-Proteinbiosynthese. Stop steht für das Stopcodon zur Beendung der Proteinbiosynthese. Nach der Transkription und Boten-RNA-Entstehung werden die Exons in dem Splice-Prozess zusammengefügt, um eine durchgehende Sequenzinformation für die Proteinbiosynthese herzustellen. Normalerweise werden die alternativen Exons nicht erkannt und die Proteinbiosynthese endet erst bei dem regulären Stopcodon in Exon 17, nachdem ein vollständiges SLP-65-Protein hergestellt wurde. Wird eins der alternativen Exons (zum Beispiel Exon 3b) erkannt, so kommt es zu einem frühzeitigen Stop der Proteinbiosynthese von SLP-65.")
Unsere Untersuchungen zeigen erstmalig, dass eine alternative oder fehlerhafte mRNA- Prozessierung zur SLP-65 Defizienz in Prä-B-ALL- Zellen führt. Warum es jedoch zu dieser veränderten mRNA Prozessierung kommt, ist zurzeit nicht bekannt und Gegenstand der aktuellen Forschung. Die Prä-B-ALL ist eine Tumorerkrankung, die in den meisten Fällen mittels einer aggressiven Chemotherapie geheilt werden kann. Trotzdem wäre es wünschenswert, mildere Therapien für diese Krankheit zu entwickeln. Wir hoffen, dass unsere weiteren Untersuchungen zur Regulation der alternativen Splice-Prozesse der SLP-65 mRNA neue Erkenntnisse zur Entstehung der B-ALL liefern und somit neue Therapieansätze für diese häufige Tumorerkrankung bei Kindern ermöglichen.